
Design Controls and Risk Management
Design controls are based on quality assurance and engineering principles. They establish a framework for manufacturers to use when designing and developing a medical device. Formal design controls must be implemented when feasibility studies are completed and a design freeze has been determined. Compliance with design controls will provide the manufacturer the opportunity to address problems early on, thereby preventing loss of time and money.
Design controls and ISO 13485 both require risk management and risk analysis to be integrated into the design process of a medical device. If performed properly, a thorough risk analysis can provide a framework for a complete validation plan for the device including specifications, performance testing, verification steps and labeling.
ISO 14971 establishes the international requirements for risk management to determine the safety of a medical device by the manufacturer during the product life cycle. The FDA’s CFR 820.30 establishes the U.S. requirements for risk management for any medical device to be sold in the U.S.
Understanding how to implement the requirements for design controls and risk management as well as how to document compliance can be a big challenge to companies seeking to market medical devices in the USA.
MDAI works with clients to produce the required set of design control documents that will show the product development process to be compliant with the international and national guidelines.
MDAI offers document development for:
- Design Input
- Requirements Specifications
- Design Descriptions
- Software Design Validation and Verification
- Risk Analysis
- Project and Verification and Validation plans
- Design Output
- Traceability Matrix
Application of Design Control Principles
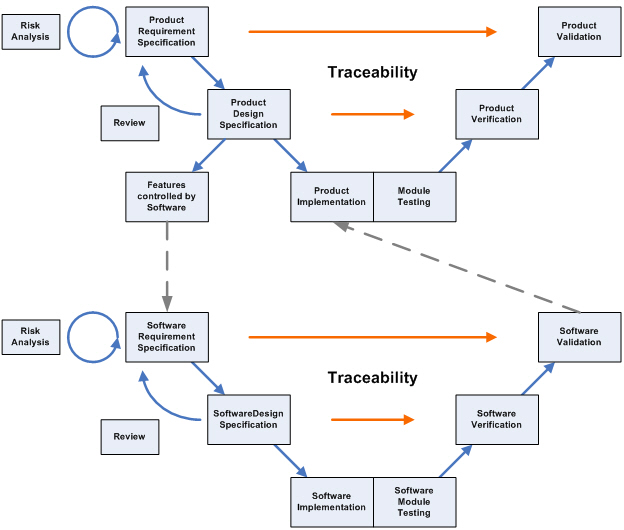
Software Validation –
The diagram above illustrates the fact that Software Validation is an integral part of the Design Control process.
“Confirmation by examination and provision of objective evidence that software specifications conform to user needs and intended uses and that the particular requirements implemented through software can be consistently fulfilled.”
(FDA guidelines)
MDAI ‘s software validation team leverages their training and experience as software engineers, their knowledge of FDA regulations for risk-based validation, and their knowledge of other similar products on the market to provide faster time to market and ultimately FDA approval without delays and questions.
Verification belongs to the software development cycle and includes checking software plans, testing the code, checking that code meets requirements and specifications. Larger manufacturers will have their own software engineers, but often the medical device manufacturer will have out-sourced the software development to an off- site IT company. In-house software development departments and off-site IT companies will have their own procedures and personnel for checking software plans, code, requirements and specifications. But not all software developers are familiar with the specific requirements of medical device software validation. Clearly the time required to obtain regulatory approval and the costs incurred can be minimized by using procedures and documentation from the beginning of the project which will be appropriate for the validation process.
Validation – Having proven that the software works without bugs and conforms to specifications the software must be validated. It must be proved to be the correct software for the device, its use must conform to safety standards, and it must be user friendly. If the requirements for validation have been attended to throughout development, then proving the validation will be swift and efficient.
Risk assessment - For Medical Device Software is crucial to the validation process.
- MDAI consultants use their knowledge of risk-based validation to appropriately scale their procedures to the complexity of the software and the risk level inherent to the device.
- Linking the risk assessment of the device to the software is efficacious in finding the failure modes inherent in the device and predicting the probability of failure that would be specific to the software.
Human factors processes are an important part of the FDA regulatory design controls. Validation should prove that the software designed conforms to the intended use of the device and takes into account the needs of the user. Many FDA recalls of medical devices can be traced to lack of utilizing user-based design principles in the development of the software. Appropriate end-user testing includes:
- Designating who will be the end-user. It could be patients, doctors, nurses, sales people.
- On site formal testing including a formal plan, data gathering and reporting, analysis of errors, understanding of risk factors and safety of the user, and gathering feedback within a real environment.
Updated or Modified Software that has not been adequately validated is the most common cause of software recalls for products already on the market. The validation for changes must be adequately tracked and attention paid to the same risk-assessment, human factors design and formal testing protocols as is required for new software.
Documentation – A well planned design for validation will ensure that the project is accurately documented in such a way as to clarify processes and action steps remaining, while producing the final documentation required for submission.
To discuss how Medical Device Approvals can help you achieve your regulatory objectives, please contact us at kathleen@mdapprovals.com